
Abstract
The purpose of this paper is to illustrate the process of differential diagnosis of the dysarthrias using an innovative case analysis approach. Using a simple acronym, the clinician integrates the available information pertaining to the patient’s etiology, lesion site, motor signs, and speech deficits to arrive at an accurate diagnosis of the type of dysarthria (or, at least rule out other types). The “hallmark” speech characteristics of each type of dysarthria will be described. In addition, the neuromuscular basis of each type of dysarthria will be presented, along with descriptions of common etiologies, sites of lesion, and non-speech motor signs. Lastly, the use of this model in the process of differential diagnosis will be demonstrated via case analyses.
Introduction
Why are there so many types of dysarthria and are they really that different from one another? What is the difference between an upper motor neuron and a lower motor neuron, and does it really matter? How can I tell if my patient’s muscles are spastic or flaccid just by listening to their speech? Why do all types of dysarthria have so many of the same speech characteristics? Why do I need to know the site of lesion in order to diagnose my patient? Can I figure it out just by listening to their speech?
These and other legitimate questions are often asked by clinicians when attempting to differentially diagnosis a patient with dysarthria. The term "dysarthria" actually refers to a group of speech disorders involving any or all of the speech production processes (i.e., respiration, phonation, resonance, articulation, and prosody) resulting from disturbances in muscular control secondary to neurological damage (e.g., degenerative diseases, cerebral vascular accidents, traumatic brain injury, etc.) (Darley, Aronson & Brown, 1975). Dysarthria is typically characterized by some degree of weakness, slowness, incoordination, or alteration of muscle tone of the speech mechanism (Duffy, 2013; Freed, 2020).
The dysarthrias are generally sub-divided into six “pure” dysarthrias (flaccid, spastic, unilateral upper motor neuron, ataxic, hypokinetic, and hyperkinetic), as well as numerous “mixed” dysarthrias (i.e., combinations of one or more pure dysarthrias) (Duffy, 2013; Freed, 2020). Each sub-type is characterized by a constellation (or “cluster”) of speech deficits, as well a corresponding neuromuscular basis. For example, spastic dysarthria results from bilateral upper motor neuron damage, which leads to spasticity of the affected muscles (i.e., hyperactive or unregulated reflexive contractions). As a result, these patients usually display speech characteristics that reflect spasticity of the affected muscles, such as harsh or strained-strangled voice, articulatory imprecision, slow speech rate, hypernasality, monopitch and monoloudness. Although patients with other types of dysarthria might also exhibit many of these same deficits, it is usually due to a different neuromuscular pathophysiology (e.g., flaccidity, ataxia, etc.). Clinically, this is a critically important distinction, as a spastic patient would typically be treated quite differently than a flaccid patient, despite several common speech characteristics. For example, strength training might be appropriate for some patients with flaccidity of the affected muscles but would be contraindicated for a patient with spastic muscles. Thus, an accurate diagnosis is crucial for effective and safe management of the patient. Simply memorizing lists of associated speech characteristics is insufficient for successful differential diagnosis of the dysarthrias (Blanchet, 2013, 2015; Dressler, Blanchet & Porcaro, 2018).
Therefore, the purpose of this paper is to explain and illustrate the process of differential diagnosis of dysarthria using the acronym ELMS (etiology, lesion site, motor signs, and speech deficits). Using this simple strategy (Blanchet, 2013, 2015; Dressler, Blanchet & Porcaro, 2018), the clinician uses all available information pertaining to these four critical aspects to aid in an accurate diagnosis of the type of dysarthria (or, to at least rule out other types). The “hallmark” speech characteristics of each type of dysarthria will be briefly described. In addition, the neuromuscular basis of each type of dysarthria will be presented, along with mention of various etiologies, sites of lesion, and non-speech motor signs. Lastly, the use of this model in arriving at an accurate diagnosis will be illustrated via case analyses.
The term dysarthria literally means “disordered utterance,” and involves impaired speech production resulting from disturbed neuromuscular control of the speech mechanism (Duffy, 2013). This distinguishes it from apraxia of speech (AOS), which, though also generally considered a motor speech disorder, involves impaired sequencing of motor commands needed to produce phonemes volitionally (Duffy, 2013). Dysarthria can involve any or all of the speech production processes (i.e., respiration, phonation, resonance, articulation, and prosody), depending on the specific type of dysarthria. Unlike AOS, which usually results from a lesion in the left cerebral hemisphere, dysarthria can result from damage in the central nervous system (CNS) or the peripheral nervous system (PNS). The sheer number of structures involved in the “motor system” (i.e., from the cerebral cortex to the muscles) helps explain why there are so many different types of dysarthria.
Five of these types were classified by Darley, Aronson, and Brown (1969a; 1969b) in their seminal Mayo Clinic study. The study involved 212 patients with one of seven categories of neurological diagnoses (e.g., parkinsonism, ALS, “palsy”). Most patients read the “Grandfather” passage, from which 30 second samples were judged by the three authors along 38 speech and voice dimensions (e.g., high pitch, short phrases, overall intelligibility, “bizarreness”). The authors then used these dimensions to create “clusters” of symptoms (e.g., the “prosodic insufficiency” cluster), or groups of symptoms that were highly correlated. This led, ultimately, to the creation of five categories of dysarthria (i.e., flaccid, spastic, ataxic, hypokinetic, and hyperkinetic). A sixth category (i.e., UUMN dysarthria) was added later, although it was alluded to by Darley, Aronson, and Brown (1969a; 1969b).
Unlike UUMN dysarthria, which was given a purely anatomic name (i.e., it is caused by a lesion to upper motor neurons on one side of the brain), the five dysarthrias categorized by Darley, Aronson, and Brown (1969a; 1969b) were given monikers that indicate the pathophysiology of the affected muscles. For example, flaccid dysarthria is caused by flaccidity of the muscles involved in speech production, which is usually caused by damage to lower motor neurons. Therefore, unless a patient’s dysarthria results from flaccidity of the affected muscles, it is not flaccid dysarthria, even though many of his or her speech deficits may be commonly associated with flaccid dysarthria. However, in my experience, neurologists’ reports do not always specify the pathophysiology or site of lesion involved using terms taught to graduate SLP students (e.g., flaccidity, spasticity, upper motor neuron, lower motor neuron, etc.).
After more than 15 years of teaching and presenting on the topic of motor speech disorders, I have discovered that this often creates confusion amongst students and clinicians attempting to diagnosis a specific type of dysarthria. Naturally, we are often tempted to “match” the observed symptoms with a list of symptoms documented to frequently co-occur in certain types of dysarthria. Unfortunately, this intuitively-appealing tactic does not always lead to an accurate diagnosis, as many speech deficits are observed in more than one type of dysarthria. In addition, a patient with a specific type of dysarthria (e.g., flaccid) might not exhibit all of the “hallmark” speech characteristics associated with that type (e.g., breathy voice quality).
Therefore, the ELMS analysis strategy (Blanchet, 2013, 2015; Dressler, Blanchet & Porcaro, 2018) was developed in an attempt to alleviate some of this confusion and to aid clinicians in the process of differential diagnosis of the dysarthrias. Using a simple written template, not unlike a S.O.A.P. note often used in hospital settings, the clinician organizes all available information related to the etiology, lesion site, motor signs, and speech deficits. Ideally, these four components should be related to each other (see Figure 1), although this is not always the case. In addition, because we do not always have all four “pieces of the puzzle,” this analytical tool assists the clinician in inferring one of the four components based on any known components, as well as his or her knowledge of the typical characteristics of each type of dysarthria. Toward that end, the four key components of each of the six “pure” dysarthrias will be reviewed and explained in the following sections. This will be followed by a demonstration of the use of the ELMS analysis via analyses of three cases modified from Duffy (2013). Prior to reading each case analysis, the reader should first become familiar with the corresponding case history (see Appendix A).
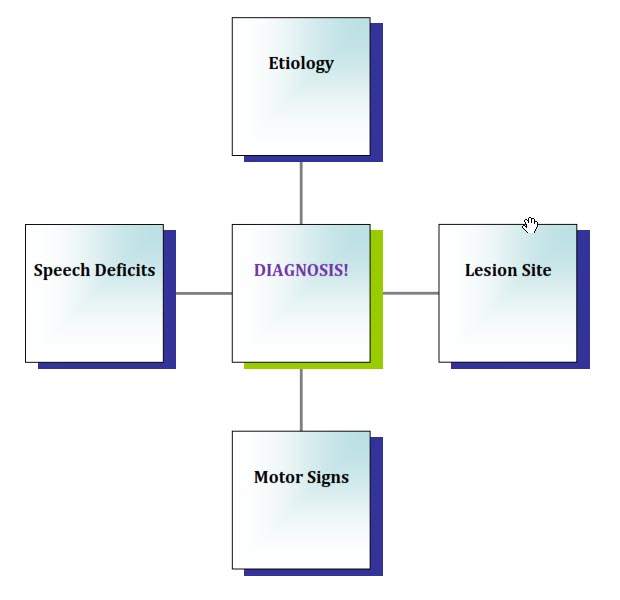
Figure 1. The four components of an ELMS analysis.
Flaccid Dysarthria
Etiology
Flaccid dysarthria (FD) results from damage to lower motor neurons (LMNs), which are simply motor neurons in the PNS (i.e., motor fibers in cranial or spinal nerves). Etiologies of FD include physical trauma, surgeries, traumatic brain injury (especially near the brainstem), and brainstem stroke (as several cranial nerves innervating head and neck muscles originate at the brainstem). A tumor can also cause FD, if it affects the brainstem or the cranial nerve itself (e.g., a neuroma). One neurological disease associated with FD is myasthenia gravis, which affects the neuromuscular junction (i.e., the point of synapse between an LMN and a muscle fiber). Patients with myasthenia gravis produce antibodies that damage acetylcholine (Ach) receptors, resulting in rapid fatigue of muscle contractions, with recovery after a rest period (Duffy, 2013). This is the only etiology of FD in which giving the patient a rest period will usually improve speech production. One useful screening task for myasthenia gravis is the “speech stress test,” wherein the clinician instructs the patient to count from 1-100 (Freed, 2020). If the patient’s voice, articulation, and/or resonance characteristics deteriorate progressively, a referral to the neurologist might be warranted.
Lesion Site
As mentioned above, FD is caused by damage to LMNs, which are motor neurons in the PNS. These fibers are also known as the “final common pathway” because they are the last fibers in the motor system to make contact with the muscles. Therefore, the neural impulse sent from the motor cortex cannot reach the intended muscle without first going through the LMNs. However, because neurology reports do not always specify “lower motor neuron” damage, it is important for the SLP to know what etiologies would be likely to cause LMN damage, as well as where these fibers are located. For example, the neurologist’s report might mention damage to a specific cranial or spinal nerve, a cranial nerve nucleus (the point at which the cranial nerve originates), or even a specific muscle (e.g., in cases of muscular dystrophy).
Motor Signs
Due to the fact that the neural impulse will not reach the muscle without going through LMNs, damage to these neurons leads to flaccidity of the affected muscles and, perhaps, “flaccid paralysis.” It is important to remember that “flaccidity” and “weakness” are not synonymous. A muscle is said to be “weak” when it has reduced ability to contract, or exhibits reduced “strength” (i.e., the ability to perform “work,” or the push or pull against an object). Flaccidity is only one cause of muscular weakness, with spasticity and rigidity being examples of others. Therefore, it is important for the SLP to check the neurologist’s report (or elsewhere in the patient’s chart) for specific signs of flaccidity. These would include muscle atrophy, fasciculations, and reduced reflexes (i.e., hyporeflexia).
Speech Deficits
Speech characteristics of FD include deficits in resonance such as hypernasality (most perceptible on vowels) and nasal emissions (most perceptible on voiceless consonants), resulting in weak pressure consonants and shortened phrases. These symptoms result from damage to CN X, the vagus nerve (specifically, the pharyngeal branch). Articulation deficits such as imprecise consonants are possible if one or more of the cranial nerves innervating the speech articulators (e.g., cranial nerves V, VII, and XII) are affected. Voice deficits such as continuous breathiness, inhalatory stridor, and harsh voice quality can result from damage to CN X (i.e., one of the laryngeal branches). Lastly, if cervical and thoracic spinal nerves are affected, the patient might exhibit respiratory deficits impacting speech production (e.g., reduced loudness, monopitch, monoloudness, harsh voice quality, shortened phrase length). Although most of these speech deficits can occur with other types of dysarthria, hypernasality, nasal emissions, and continuous breathiness are most often seen in cases of flaccid dysarthria. Spastic dysarthria is the only other type which often involves severe hypernasality, although usually without the nasal emissions observed in cases of FD. Hypokinetic dysarthria is the only other type that often presents with continuous breathy voice quality (Blanchet & Snyder, 2009).
Spastic Dysarthria
Etiology
The neurological basis of spastic dysarthria (SD) is bilateral damage to upper motor neurons (UMNs), which are simply motor neurons in the CNS (i.e., the brain and spinal cord). These fibers are “bundled” together in groups called tracts (similar to nerves in the PNS), some of which serve muscles of speech production. The most common etiology of bilateral UMN damage is a stroke, either a single stroke in the brainstem (where left and right motor tracts course closely together), or a single cortical stroke following one in the other hemisphere. Other etiologies of SD include traumatic brain injury (TBI), brainstem tumors (when they compress UMNs or cranial nerve nuclei), cerebral anoxia, or infections (e.g., meningitis).
Lesion Site
As mentioned above, SD results from bilateral damage to UMNs, specifically the pyramidal and extrapyramidal tracts that serve speech muscles (Duffy, 2013). Fibers in the pyramidal system (specifically, the corticobulbar tracts) originate in motor cortex and synapse with LMNs at the brainstem. The term “bulbar” means “pertaining to the brainstem,” hence the name “corticobulbar tract.” The majority of pyramidal fibers decussate (i.e., cross) at the pyramids of the medulla, hence the name “pyramidal” system. These tracts are also known as the “direct activation pathway,” as they send the neural signal down to the LMNs to initiate volitional movements of selected muscles. The extrapyramidal system consists of fibers originating in cortex and brainstem, which make numerous connections before eventually synapsing with LMNs. This system is also known as the “indirect activation pathway.” The extrapyramidal system helps to maintain posture, regulate reflexes, and monitor muscle tone; it works in parallel with the pyramidal system (Duffy, 2013).
Motor Signs
As bilateral damage to UMNs usually involves both pyramidal and extrapyramidal tracts, this damage usually results in a combination of symptoms, including weakness, slow movements, spasticity, and hyperreflexia. The spasticity experienced by these patients is due to damage to extrapyramidal fibers, which help regulate and control our reflexive movements when we do not want them to occur (for example, when you pick up a hot cup of coffee, but want to carry it to the table rather than drop it on the floor). Unless they are inhibited by the motor cortex, the patient’s reflexive contractions eventually predominate, resulting in spasticity (which is simply unregulated reflexive contraction). Additional characteristics to look for in the neurologist’s report include pseudobulbar affect (i.e., “emotionally lability”) and the return of “primitive” reflexes, such as the Babinski sign, suckle reflex, snout reflex, and jaw jerk reflex. These symptoms often occur following UMN damage, as the cerebral cortex is no longer able to effectively inhibit these behaviors that are normally inhibited in adults.
